Inner ear malformations (IEM) represent about 20%–35% of the etiology of congenital sensorineural hearing loss (SNHL).
Incomplete cochlear partition anomalies characterize a group of IEM with normal cochlear location, external dimensions (height and length) but with various internal architecture defects. In these collections of malformations, there is a clear distinction between a cochlea and vestibule.
According to the latest classification of cochleovestibular malformations by Sennaroglu and Saatci, based on the defect in the modiolus and the interscalar septa there exists three different types of incomplete partition groups, namely.
- Incomplete cochlear partition Type I
- Incomplete cochlear partition Type II (Classic Mondini deformity)
- Incomplete cochlear partition Type III
Incomplete Cochlear Partition type I (IP-I)
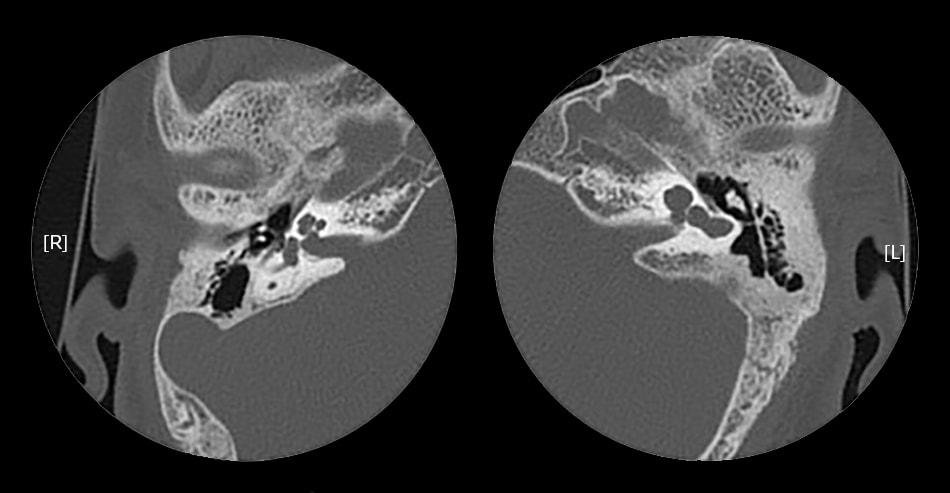
This type was termed as “cystic cochleovestibular malformation” in 2002 by Sennaroglu and Saatci. The cochlea is lacking the entire modiolus and cribriform area, resulting in a cystic appearance and is accompanied by a large cystic vestibule.
This group represents approximately 20% of inner ear malformations.
Pathophysiology of IP-1
Studies have shown that IP-1 defect occurs due to the arrest of embryological development, usually in the early 5th week of life, so that the cochlea and vestibule can be differentiated, but they have no internal architecture and appear as empty cystic spaces.
Recent histopathological studies suggest that IP-I may be due to endosteal developmental abnormality as a result of defective vascular supply coming from the Internal Acoustic Canal (IAC). This can also result in abnormal/malformed stapes footplate also.
Radiological characteristics
The cochlea will be located in its normal location, in the anterolateral part of the fundus of the IAC and is having normal external anatomy.
IP-I cochlea lacks the entire modiolus and interscalar septa, making it appear like an empty cystic structure. It is accompanied by an enlarged and dilated vestibule. Vestibular aqueduct enlargement is usually rare.
All patients of IP-1 will have an enlarged Internal Acoustic Canal. There may be a defect between the IAC and the cochlea due to the developmental abnormality of the cochlear aperture and the absence of the modiolus, and CSF may completely fill the cochlea.
Recurrent meningitis in Incomplete Cochlear Partition-1
Recurrent meningitis can occur in IP-I patients. The high CSF pressure filling the cochlea disrupts the thin (sometimes malformed) stapes footplate, leading to a CSF fistula at the oval window causing meningitis during an attack of otitis media.
All patients with IP-I and recurrent meningitis with normal tympanic membranes but fluid filling the middle ear and mastoid should have an exploration of the middle ear with special attention to the stapes footplate.
Cochlear implantation in IP-I
Majority of Incomplete Cochlear Partition-I patients will have severe to profound sensory neural hearing loss and gain little benefit from traditional hearing aids; hence a majority of them are candidates for cochlear implantation (CI). Some are associated with cochlear nerve aplasia, which excludes then from the cochlear implantation candidacy. Auditory brain stem implantation can be considered for such candidates.
Cochlear implantation in patients with IP-I may be challenging for the surgeon due to various reasons.
Facial nerve abnormalities can also be seen in IP-I because of the associated enlarged vestibule and anomalous lateral semicircular canal. This limits the classical transmastoid- facial recess and an alternative transcanal approach can be used as reported by Sennaroglu (2010). For the same reason, facial nerve monitoring should be considered in all CI surgery in the malformed inner ear.
As the modiolus is entirely lacking, and the exact location of the ganglion cells is not precisely known, full-banded straight electrodes may be more appropriate. Since the size of the cochlea is normal, straight electrode about 25 mm is preferred.
During CI surgery gusher is very common (40-50%) which necessitates special precautions. If the cochleostomy is not properly sealed, this may lead to a CSF fistula with recurrent meningitis. The cork / FORM 24 (Med El) electrodes designed by Sennaroglu (2010) can be considered in these cases. Digisonic Classic and Digisonic Evo electrodes (Oticon) also have a silicon stopper which might be useful for controlling gusher.
Results of CI in IP-1 are variable and are mainly related to the residual neural nerve fibers and surgical placement of the electrode. The lack of modiolus results in a very poor residual neural activity which limits the surgical outcome. But in many cases the results are satisfactory.
References
- Ruhl DS, Koehn HA, Mahaney KB, Kesser BW. Congenital Fistulae of the Stapedial Footplate and Round Window Membrane: An Unusual Cause of Recurrent Meningitis. JAMA Otolaryngology–Head & Neck Surgery. 2018 Jan 1;144(1):89-90.
- Sennaroglu L, Saatci I. A new classification for cochleovestibular malformations. Laryngoscope. 2002;112 (12): 2230-41
- Sennaroglu L, Sarac S, Ergin T. Surgical results of cochlear implantation in malformed cochlea. Otol Neurotol. 2006;27:615–623.
- Sennaroglu L. Cochlear implantation in inner ear malformations – a review article. Cochlear Implants Int. 2010;11:4–41.
- Berrettini S, Forli F, De Vito A, Bruschini L, Quaranta N. Cochlear implant in incomplete partition type I. ACTA otorhinolaryngologica Italica. 2013 Feb;33(1):56.
- SennaroÄŸlu L, Bajin MD. Classification and Current Management of Inner Ear Malformations. Balkan medical journal. 2017 Sep;34(5):397.