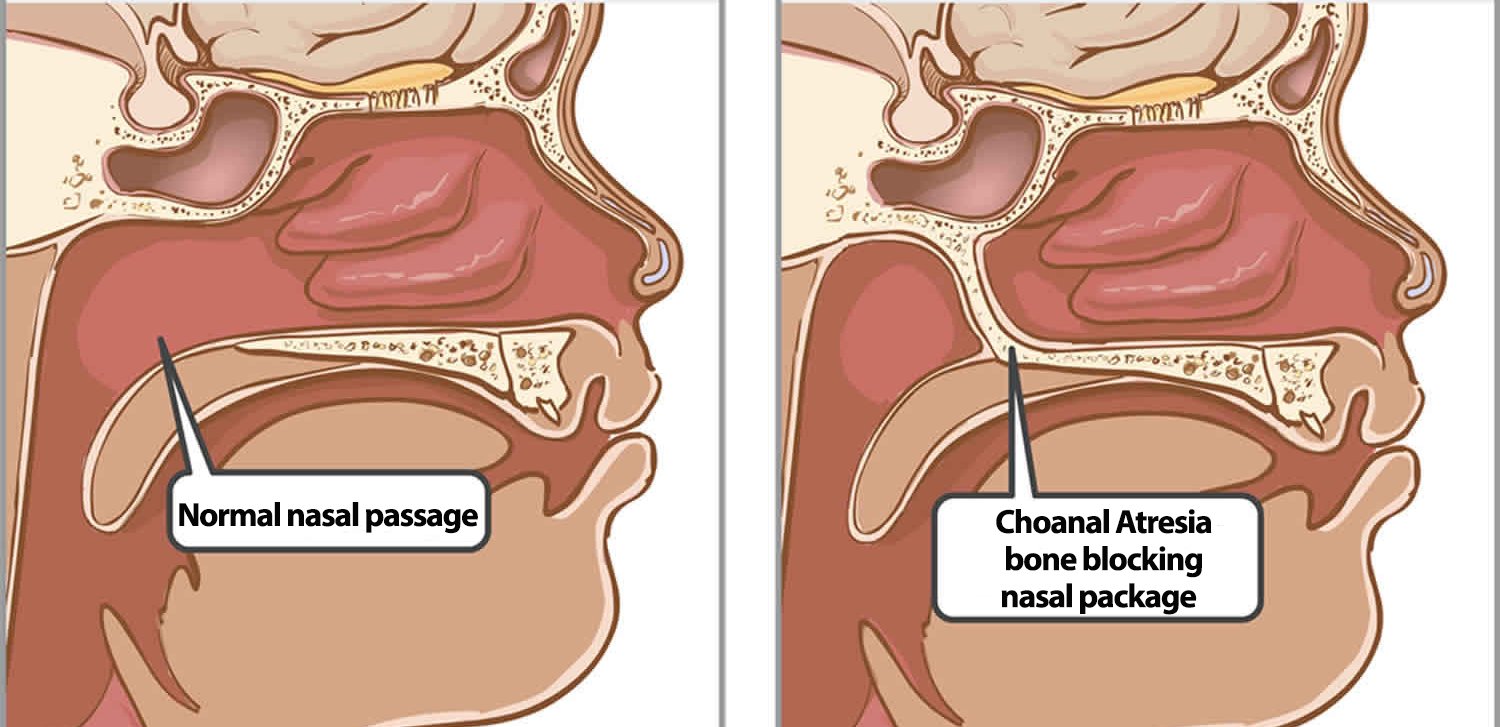
Choanal atresia is a rare congenital abnormality with a reported incidence of 1 in 5000 / 8000 live births.
The condition is characterized by the partial or complete obstruction of the nasal airway at the level of the choanae. It is a rare condition that occurs due to a failure of the normal development of the nasal passages during embryogenesis.
The condition can be unilateral or bilateral and may result in respiratory distress and feeding difficulties in affected infants. Understanding the embryology, anatomy, clinical features, diagnosis, evaluation, and treatment of choanal atresia is essential for proper management and improved patient outcomes.
Anatomy
The nasal cavity is divided into two main parts: the anterior nasal vestibule and the posterior nasal cavity. The posterior nasal cavity communicates with the nasopharynx through paired openings – the choanae.
Embryology
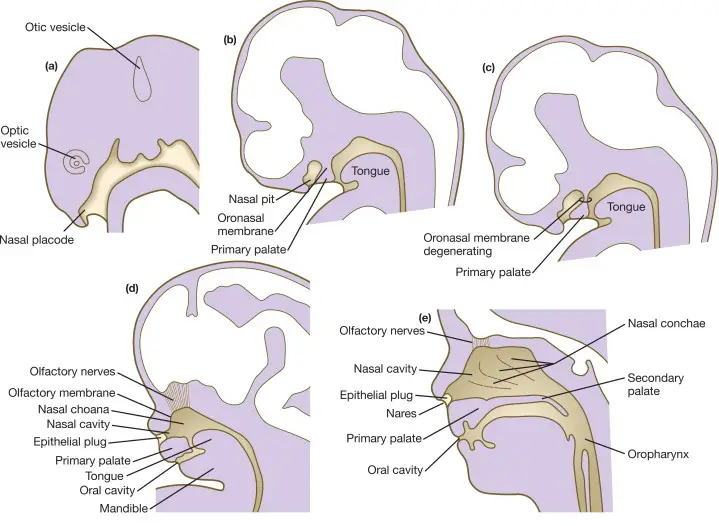
During embryonic development, the nasal cavities originate from the nasal pits, which are invaginations in the facial ectoderm. The nasal pits deepen to form the primitive nasal sacs, which subsequently grow dorsally and medially. During this time, the oronasal membrane (which forms the palate later) separates the nasal cavity from the oral cavity.
Between the third and seventh embryonic weeks, these sacs eventually fuse with the developing oronasal membrane. Failure of this fusion process can result in choanal atresia, where the choanae, the posterior openings of the nasal cavities, do not develop properly.
Classifications
- Unilateral – Unilateral choanal atresia cases usually remain unnoticed until adult life. Presence of unilateral mucoid discharge devoid of air bubbles is the usual clinical presentation.
- Bilateral – 40% cases are bilateral. Since infants are obligate nose breathers, bilateral choanal atresia is a life-threatening emergency, causing hypoxia, apnoea and failure to thrive.
- Complete or Incomplete
- Bony (90%) or membranous (10%) – bony choanal atresia, which involves the bony portion of the choanae, and membranous choanal atresia, where the blockage occurs at the membranous portion.
Clinical Features
Choanal atresia is more often unilateral than bilateral (60% vs. 40%) and occurs more frequently in females than in males (ratio 2:1).
The clinical presentation of choanal atresia varies depending on the severity and laterality of the obstruction.
Unilateral atresia may be less symptomatic, with one nasal passage being open and allowing some airflow. However, these infants may still experience recurrent nasal obstruction, non resolving purulent nasal discharge on the affected side, chronic congestion, and sinus infections. Feeding difficulties, including poor sucking and choking during feeds, may also be observed.
Newborns with bilateral atresia typically present with severe respiratory distress immediately after birth. They may have difficulty breathing through the nose and may exhibit nasal flaring, cyanosis (bluish discoloration), and retractions (pulling in of the chest wall). The cyanosis is relieved with crying and returns with rest (paradoxical cyanosis). Since newborns are obligate nasal breathers, establishing an airway in bilateral atresia at birth is an acute otolaryngologic emergency.
Bilateral atresia, affecting both sides, is often associated with syndromic conditions.
Diagnosis
The diagnosis of this condition should be done immediately after birth. The initial clinical evaluation includes placing a laryngeal mirror under the nostril to check for fogging and introducing a suction catheter through each nostril and into the child’s oral cavity.
Failure to pass a suction catheter from nose to pharynx usually hints the diagnosis. Other tests though not done these days are
- Drops of methylene blues instilled to nose won’t come to pharynx.
- Xray lateral view after putting radio-opaque dye into nose will show atresia.
Anterior rhinoscopy, performed using a flexible or rigid nasal endoscope, can aid in visualizing the atretic plate, which obstructs the choanae.
Imaging studies, such as computed tomography (CT) scans, are valuable for assessing the choanal anatomy, identifying the specific site of atresia, and detecting any associated abnormalities or syndromes. CT is also helps in differentiating other causes of nasal obstruction from choanal atresia.
Differential diagnoses
Differential diagnoses include
- pyriform aperture stenosis,
- nasolacrimal duct cysts,
- chordoma,
- turbinate hypertrophy,
- septal dislocation and deviation,
- antrochoanal polyp, or
- nasal neoplasm.
Evaluation of Choanal atresia
In addition to clinical and imaging evaluations, further assessments may be warranted. A complete blood count may be performed to evaluate for signs of infection or anemia. Blood gas analysis helps assess the severity of respiratory distress and the need for intervention.
Genetic testing may be recommended to identify any underlying genetic abnormalities or syndromes associated with choanal atresia.
It is important to evaluate for any associated congenital anomalies, such as cardiac defects, craniofacial abnormalities, or genitourinary anomalies, as these can impact management decisions and overall prognosis.
Syndromic associations
Choanal atresia can be associated with various syndromes. Some of the commonly observed syndromic associations include:
- CHARGE syndrome: This is a complex genetic disorder characterized by a constellation of features, including coloboma (eye abnormality), heart defects, choanal atresia, retardation of growth and development, genitourinary abnormalities, and ear abnormalities.
- Treacher Collins syndrome: It is a craniofacial disorder characterized by underdeveloped facial bones, downward-slanting eyes, absent or abnormal ears, and cleft palate. Choanal atresia can be present in some individuals with this syndrome.
- DiGeorge syndrome (22q11.2 deletion syndrome): This genetic disorder involves the deletion of a small piece of chromosome 22. It is associated with a wide range of features, including heart defects, immune system abnormalities, characteristic facial features, developmental delay, and choanal atresia.
- Fraser syndrome: This rare genetic disorder is characterized by abnormalities in multiple systems, including the eyes (cryptophthalmos), ears, respiratory tract (choanal atresia), kidneys, and genitalia. It is caused by mutations in the FRAS1 gene.
- Opitz G/BBB syndrome: This is a genetic disorder characterized by midline defects, including choanal atresia, cleft lip and/or palate, heart defects, and developmental delays. It can have variable expressivity and is caused by mutations in various genes involved in midline development.
It is important to note that these syndromic associations are not exclusive to choanal atresia and can occur in combination with other congenital anomalies. A thorough evaluation and genetic testing may be recommended to identify associated syndromes and guide appropriate management and counseling for affected individuals and their families.
Treatment of choanal atresia
Treatment of choanal atresia is essentially surgical. The primary goal of treatment for choanal atresia is to establish a patent nasal airway.
As a temporary solution, McGovern’s technique can be considered – providing the kid with a feeding nipple having a large hole which can obviate the need for endotracheal intubation or tracheostomy – provides good oral airway. The atresia plate is later surgically corrected.
Unilateral choanal atresia does not require surgical treatment as urgently as the bilateral case, and can be postponed until school-age when the anatomy of the region is more similar to that in adults.
Immediate intervention is necessary for infants with bilateral atresia to ensure adequate respiration. Surgical repair is the mainstay of treatment and is typically performed within the first few weeks of life. The choice of surgical technique depends on various factors, including the anatomy of the atresia, the surgeon’s expertise, and associated anomalies.
Two common approaches include the endonasal approach, where the blockage is accessed through the nostrils, and the transpalatal approach, where access is gained through the roof of the mouth. The surgical repair involves removing the obstructing plate and reconstructing the choanae to create a functioning nasal airway.
The endoscopic endonasal technique should be considered the first choice for the surgical treatment of choanal atresia as it offers a direct approach to the atretic plate, reduces intraoperative bleeding, reduces hospitalization time, and lowers morbidity.
The use of choanal stenting and mitomycin C as an adjunct therapy to prevent restenosis are a controversial topic in the management of choanal atresia as there is no clear-cut evidence on the effectiveness of using stents or mitomycin after choanal atresia repair.
Prognosis
The prognosis for choanal atresia is generally favorable, especially with early diagnosis and prompt surgical intervention.
The factors influencing treatment outcomes include the experience of the surgical team, the anatomy and type of atresia, the presence of associated anomalies, and any underlying syndromes. Complications can occur, albeit infrequently, and may include restenosis (narrowing of the choanae), nasal adhesions, infection, bleeding, or injury to adjacent structures.
Long-term follow-up is essential to monitor the patient’s nasal airway function, growth and development, and potential recurrence or late complications. In some cases, additional surgeries or interventions may be required to address any residual issues or complications.
It is important to note that the prognosis and treatment outcomes can vary among individual patients. Multidisciplinary care involving otolaryngologists, neonatologists, geneticists, and other specialists is often necessary to provide comprehensive management and optimize outcomes.
Understanding the embryology, anatomy, clinical features, diagnosis, evaluation, and treatment options is crucial for healthcare professionals involved in the care of affected infants. Further research is needed to optimize surgical techniques and long-term outcomes for individuals with choanal atresia.
References
- Sajan J, et al. (2017). Congenital nasal pyriform aperture stenosis. StatPearls. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK499877/
- Mitchell RB. (2017). Choanal atresia: Diagnosis and management. Curr Opin Otolaryngol Head Neck Surg, 25(6), 509-513.
- Sidell DR, et al. (2018). Choanal atresia and choanal stenosis. Otolaryngol Clin North Am, 51(6), 1091-1107.
- Cofer SA, et al. (2020). Choanal atresia repair: A review of techniques, outcomes, and complications. Int J Pediatr Otorhinolaryngol, 128, 109699.
- Development of face – https://doi.org/10.1016/B978-0-12-801238-3.05451-9